
Abstract
Background: Laurel wilt disease has caused the extensive mortality of lauraceous species in the southeastern United States. The causal agent is an invasive fungus, Raffaelea lauricola, which is a symbiont of the beetle Xyleborus glabratus and causes a rapid, fatal vascular wilt. Early diagnosis of laurel wilt is imperative for efficient disease management. The current diagnostic process, however, is slow due to the lengthy laboratory procedures required to confirm pathogen presence. Methods: We tested the robustness and field-portability of a recently developed, species-specific, loop-mediated isothermal amplification (LAMP) assay for R. lauricola, with the overall goal of eliminating the need for a laboratory confirmation of the diagnosis. We tested the robustness of the assay using benchtop equipment with naturally infected samples. We then tested the assay directly in the field using a portable device. Results: The assay successfully detected R. lauricola directly from symptomatic wood tissue using crude DNA extracts. Furthermore, the assay readily allowed users to distinguish between symptoms caused by R. lauricola infection and similar symptoms caused by other agents. In-field, we assayed wood samples from symptomatic redbay (Persea borbonia [L.] Spreng) and sassafras (Sassafras albidum [Nutt.] Nees) across the Southeast and successfully detected R. lauricola-infected trees in less than an hour. Conclusion: Results of this study confirmed that the field-deployable LAMP assay is robust and can rapidly and accurately detect R. lauricola in infected trees directly on-site. LAMP technology is well suited for in-field implementation, and these results serve as an incentive for further development and use of this technology in the field of forest pathology.
- Crude DNA
- Early Detection and Rapid Response
- In-Field
- Loop-Mediated Isothermal Amplification (LAMP)
- Raffaelea lauricola
INTRODUCTION
Invasive diseases are one of the greatest threats to forests and individual trees, and pathogenic fungi and fungus-like organisms in particular cause the greatest amount of damage (Santini et al. 2013), such as in the case of the pathogens that cause chestnut blight and Dutch elm disease, each of which has resulted in the deaths of millions of trees (Freinkel 2007; Martín et al. 2018). In addition to their devastating effects on ecosystems, the cost of managing invasive species can reach into the billions of dollars annually (Pimentel et al. 2005; Aukema et al. 2011; National Invasive Species Council 2016). Efforts to protect trees from invasive fungal diseases are paramount to ensure the preservation of the benefits they provide. Early detection, followed by a rapid response, are among the crucial steps of frontline defense to contain invasive species and mitigate their damage (Lamarche et al. 2015; National Invasive Species Council 2016; Eschen et al. 2018).
Laurel wilt is an invasive, fatal vascular disease that has resulted in extensive mortality among North American members of the Lauraceae family (Fraedrich et al. 2008). The species most commonly affected are redbay (Persea borbonia [L.] Spreng), sassafras (Sassafras albidum [Nutt.] Nees), and avocado (Persea americana Mill.), which collectively include ecologically and economically important forest, urban, and agriculture trees (Fraedrich et al. 2008; Ploetz et al. 2012). The disease is caused by an ambrosia fungus, Raffaelea lauricola, which is carried by a beetle vector, Xyleborus glabratus (redbay ambrosia beetle) (Fraedrich 2008; Harrington et al. 2008). Although R. lauricola is a secondary pest of dying and unhealthy trees in its native Asian range (Shih et al. 2018), in North America the fungus is extremely pathogenic to many native lauraceous species (Hughes et al. 2015), including those currently outside of impacted areas, such as California laurel (Umbellularia californica [Hook. & Arn.] Nutt.)(e.g., Fraedrich 2008). Upon being introduced to Georgia and South Carolina by 2002 (Fraedrich et al. 2008), laurel wilt disease quickly spread south throughout Florida, west to Texas (Menard et al. 2016), along the Atlantic coast up to North Carolina (North Carolina Forest Service 2012), and has recently crossed the Appalachian Mountains into Tennessee and Kentucky (Loyd et al. 2020).
Since the disease is already widely established in the southeastern United States, eradication efforts are no longer viable, and current management suggestions include early detection and removal of infected trees in an effort to slow the spread of laurel wilt disease to new areas (Hughes et al. 2015). Early detection, in particular, is the most crucial and currently the most inefficient step for the successful management of the disease (Hughes et al. 2015). The guidelines in place to confirm the disease involve a lengthy process, including the shipment of a suspected sample to an equipped laboratory and the isolation of a pure fungal culture, followed by DNA extraction and species-specific PCR (Hughes et al. 2015). This is a time intensive process, often taking a week or more, and it significantly delays any disease management options. Furthermore, there are currently no PCR protocols that can reliably detect R. lauricola directly from host tissues, thus necessitating the isolation of the fungus before any molecular test can be performed (Dreaden et al. 2014) and emphasizing the need for a diagnostic test that can quickly and accurately detect R. lauricola directly from host tissues, ideally in the field.
Loop-mediated isothermal amplification (LAMP) is a relatively recent molecular technique (Notomi et al. 2000) that has the capability of providing point-of-care diagnostic testing (Niessen 2015). LAMP is less sensitive to inhibitors than PCR (Francois et al. 2011), allowing for the use of crude DNA extracts, which can be rapidly obtained under field conditions (Kogovsek et al. 2015; Colombari et al. 2016). LAMP reactions are also isothermal, thereby removing the requirement for a thermal cycler (Notomi et al. 2000). While basic LAMP reactions can be performed on a simple hot plate, portable devices that feature a heating element along with fluorescence-based detection are also available for real-time monitoring (Ebert et al. 2010; Jenkins et al. 2011). The use of portable LAMP devices fulfills the criteria for Early Detection and Rapid Response as outlined by the US Department of the Interior for safeguarding natural resources (US Department of the Interior 2016) and would be a useful tool for the rapid detection of invasive pathogens such as R. lauricola. LAMP assays have been shown to successfully diagnose plant diseases, including those caused by bacterial (e.g., Keremane et al. 2015; Ocenar et al. 2019), viral (e.g., Congdon et al. 2019), and fungal pathogens (e.g., Tomlinson et al. 2010; Villari et al. 2013; Aglietti et al. 2019).
We recently designed a species-specific LAMP assay for the molecular detection of R. lauricola directly from host tissues in less than 20 minutes of reaction time and as early as 12 days after inoculation when using crude DNA extracts (Hamilton et al. 2020). However, the assay has been only tested with artificially inoculated and otherwise clean redbay samples, and only on a benchtop instrument in a laboratory setting. Further testing is needed to ensure the approach is suitable when used in the field, when samples might be colonized by multiple organisms, and when laboratory equipment is not available. Building upon our previous work, the aim of this study was to evaluate the performance of the recently designed R. lauricola species-specific LAMP assay for use under field conditions comparable to those actually encountered by laurel wilt surveillance personnel, arborists, or orchard managers. With respect to this, we divided the study into 2 successive phases with the specific objectives of (I) testing the suitability of the LAMP assay when using crude DNA extracts from naturally infected sassafras and redbay wood samples in a laboratory setting, and (II) verifying the feasibility of the LAMP assay when conducted directly in the field on portable equipment.
MATERIALS AND METHODS
Objective I: Testing of Naturally Infected Samples
Sample Collection
Redbay and sassafras samples were collected in the summer and autumn of 2018, respectively, from 6 different locations in 6 different states of the southeastern United States, including Arkansas, Georgia, Louisiana, North Carolina, South Carolina, and Texas. At each location, 2 sites were selected: one where laurel wilt disease was known to occur, and one where the disease did not occur. From each site with diseased trees, 5 symptomatic and 5 non-symptomatic trees were sampled (Table 1), while at sites with no disease, only 5 non-symptomatic trees were sampled (Table 2). The only exceptions to this sampling design were that (1) in South Carolina, no non-diseased site could be found, and (2) at the Louisiana location, only 3 trees were sampled per symptom type per site. Samples consisted of 10-cm-long branch or stem sections, ranging between 4 mm and 15 mm in diameter. Sampling was accomplished with the assistance of local state and federal forestry agencies, who identified and collected the samples during their routine surveillance operations and then shipped them overnight on ice to the University of Georgia. Once received, samples were stored at −20 °C until further processing and analyses.
Results of laboratory LAMP assays and corresponding validation via fungal isolation to detect Raffaelea lauricola from wood tissue samples collected from sites with laurel wilt disease in the southeastern US. Results are reported as number of positive assays or fungal isolations out of the total samples tested.
Results of laboratory LAMP assays and corresponding validation via fungal isolation to detect Raffaelea lauricola from wood tissue samples collected from sites without laurel wilt disease in the southeastern US. Results are reported as number of positive assays or fungal isolations out of the total samples tested.
DNA Extraction in the Laboratory
Crude DNA extracts were obtained following the protocol described in Hamilton et al. (2020). Briefly, 15 mg to 20 mg of wood shavings were removed from samples with a scalpel after debarking and added to a 1.5 mL micro-centrifuge tube containing 300 mL of 5% Chelex 100 (BioRad, California, USA). The wood shavings were then boiled for 5 minutes, vortexed for 15 seconds, boiled again for 5 minutes, and vortexed again for 15 seconds, before being spun down in a mini-centrifuge (VWR, Radnor, PA, USA) at 3,884 × g for 30 seconds. The supernatant was used as template in the LAMP reactions.
LAMP Assay on a Benchtop Instrument
LAMP reactions were performed as per Hamilton et al. (2020) with one modification, whereby less DNA template (i.e., the total DNA that goes in every reaction) was used in order to reduce background noise and potential inhibition of the LAMP assays. Each 25 μL reaction contained 15 μL 1× no-dye Isothermal Master Mix (Optigene, Horsham, UK), 0.7 μL of each internal primer BT-FIP and BT-BIP (100 μM), 0.7 μL of each external primer BT-F3 and BT-B3 (10 μM), 0.2 μL of F-Loop primer (100 μM), 0.24 μL of assimilating probe fluorescent (FAM) strand (10 μM), 0.46 μL of quencher strand (10 μM), 5.3 μL of molecular grade water (Thermo Fisher Scientific, Waltham, MA, USA), and 1 μL of crude DNA template. All primers were diluted in molecular grade water. Reactions were carried out in 0.2-mL optically clear PCR eight-tube strips (Thermo Fisher Scientific) and run on a benchtop StepOnePlus Real-Time PCR system (Thermo Fisher Scientific) programmed with the following conditions: preheat to 65 °C for 1 minute, 65 °C for 30 minutes with fluorescence reading every minute, followed by a denaturing step at 85 °C for 5 minutes to halt the reaction and deactivate the polymerase. Each sample was run in triplicate, and each reaction included a tube with 1 ng of high quality DNA of R. lauricola isolate FS-0009 (Hamilton et al. 2020) as a positive control, in addition to a no-template negative control. A result was considered positive if 2 out of 3 technical replicates amplified.
Fungal Isolation
To verify the presence of the pathogen in each tested sample, a portion of tissue of each sample was plated onto amended malt extract agar. Processing consisted of debarking the sample and surface sterilizing it by dipping it in 10% common bleach in water (corresponding to 8.25% sodium hypochlorite) for 10 seconds, then 70% EtOH for 10 seconds, and finally in sterile water for 10 seconds. After the sample was allowed to air dry, it was placed onto malt extract agar (VWR) amended with 200 ppm cycloheximide (Sigma-Aldrich, St. Louis, MO, USA) and 100 ppm streptomycin (Sigma-Aldrich)(CSMA)(Harrington and Fraedrich 2010). Plates were wrapped with parafilm (Bemis, Neenah, WI, USA) and incubated at 25 °C in an Isotemp® incubator (Model 655D, Thermo Fisher Scientific) until R. lauricola growth was observed or a minimum of 2 weeks had passed. Identification of R. lauricola was performed based on morphological features such as its characteristic mucoid growth, the presence of conidiophores, and the shape and size of its conidia (Harrington et al. 2008).
Objective II: In-Field Testing of the LAMP Assay
Sample Collection
Redbay and sassafras samples were assayed in-field from November 2019 to March 2020 in 5 different southeastern states, including Georgia, Kentucky, North Carolina, South Carolina, and Tennessee (Table 3). As previously noted, site locating and sampling was again achieved with the assistance of local state and federal forestry agencies. At each location, testing was performed at a single site on a single day, with the exception of Georgia, where redbay trees were sampled at one site and sassafras trees at another, but the testing was still carried out on the same day. At least 6 individual potentially diseased trees were selected in each state based on visual symptoms, including leaf wilt and sapwood discoloration upon debarking. Collected samples consisted of sapwood tissue from the main stem of larger trees or entire sections of stems from sprouts and saplings.
Results of the in-field LAMP assays and corresponding laboratory verification to detect Raffaelea lauricola from wood tissue samples collected and tested in-field at sites with laurel wilt disease in the southeastern US.
DNA Extraction in the Field
For in-field crude DNA extractions, we used a protocol similar to that previously used in the laboratory, but with some adaptations to accommodate the field conditions. In particular, we wanted the protocol to be workable with minimal equipment and to avoid an open flame. Stem wood samples were acquired from visually symptomatic host trees only. For smaller diameter trees, a hand saw was used to fell trees and expose discolored sapwood, and whole stem sections were then collected. For larger diameter trees that could not be readily felled, a hatchet was used to remove bark and expose discolored sapwood, and samples were collected with a pocket knife. Tools were disinfected with 10% bleach and 70% ethanol in between each sample. From each tree, part of the collected wood sample was processed for crude DNA extraction and LAMP analysis directly on-site, while another part was stored in a clean resealable plastic bag and transported in a cooler with ice packs back to the laboratory for further analyses. One to three thinly shaved, discolored pieces of sapwood (comparable in terms of weight to the 15 mg to 20 mg used for the crude extraction protocol in the laboratory) were placed into a previously prepared extraction tube containing 300 mL of 5% Chelex 100. When the wood discoloration was particularly dark, approximately half as much wood was used in the extraction to avoid excessive background noise and potential inhibition of the LAMP assay. Extraction tubes were then placed in a floating tube rack and boiled in a low wattage 110-V, 0.5-L electric kettle (DCIGNA190327, DCIGNA, Wuhan, China). The kettle was either plugged into an electricity supply when available (e.g., at the state forestry building nearby the sampling location in Tennessee) or into a 1500-W power inverter (Model #HD1500, Krieger, Fort Lauderdale, FL, USA) connected to the battery of a running vehicle. The lid was removed from the kettle to prevent accumulation of vapor, which would trigger its turning off. Samples were boiled for 5 minutes, manually shaken for 10 seconds, boiled for an additional 5 minutes, shaken again, and then stored in a cooler with ice packs until testing. Prior to testing, samples were centrifuged 2,000 × g for 10 seconds on a Corning LSE #6770 mini-centrifuge (Corning, NY, USA) connected to the same power source previously used for the kettle, and only the supernatant was used as template in the LAMP reactions.
LAMP Assay on a Portable Device
Preliminary testing was performed using a BioRanger™ Platform (Diagenetix, Honolulu, HI, USA), but because the device functioning was not consistent (data not shown), we decided to proceed with a Genie® III (Optigene) portable LAMP device. LAMP reactions in the field were performed using the same crude DNA reaction mix as described above, which had been prepared beforehand (24 hours to 36 hours prior to testing) and already aliquoted into strips. Ready-to-use reaction strips, with each including a positive control and a negative control as described above, were double wrapped in aluminum foil to avoid photodegradation of the probes and stored at 4 °C until use. During travel, reagents were constantly kept on ice. After adding 1 μL of template, LAMP reactions were performed on the Genie® III (Optigene) device programmed with the following conditions: 65 °C for 30 minutes with the default fluorescence reading (15 seconds) followed by an inactivation step at 85 °C for 5 minutes to halt the reaction and deactivate the polymerase.
Verification of Field Results
Field collected samples were transported to the laboratory to further verify the presence of R. lauricola. A portion of tissue of each sample was plated onto amended malt extract agar as previously described, and the presence of mycelial growth of R. lauricola or its absence was recorded. Discrepancies among field LAMP assay results and plating were further assessed as follows: (1) if a sample tested positive according to the in-field LAMP assay, but R. lauricola was not recovered when plated, a high quality DNA extraction followed by a LAMP assay on the benchtop equipment was performed following the protocol previously described; (2) if a sample tested negative according to the in-field LAMP assay, but R. lauricola was recovered when plated, a crude DNA extraction with less starting material, to reduce potential inhibition, was performed in the laboratory, followed by a LAMP assay on the portable device, as previously described. For high quality DNA extractions, samples were ground in liquid nitrogen and extracted with a Qiagen DNeasy plant mini kit (QIAGEN, Germantown, MD, USA) as per the manufacturer’s protocol.
Statistical Analysis
To determine the accuracy of the in-field LAMP assay compared to the actual presence of R. lauricola in the samples, the true positive rate (i.e., sensitivity) was calculated as the number of correct positive predictions (i.e., positive in-field LAMP assays confirmed by either laboratory isolations or high quality DNA testing) divided by the total number of positives (i.e., positive laboratory isolations and high quality DNA testing)(Yerushalmy 1947) using Microsoft Excel® 2013 (Version 15.0.5215.1000).
RESULTS
Objective I: Testing of Naturally Infected Samples
LAMP Testing in the Laboratory
The results of the laboratory validation of naturally infected samples collected in diseased locations are reported in Table 1, whereas the samples collected in the non-diseased locations are reported in Table 2. Of the symptomatic host samples received from cooperators, 4 out of 5 samples from North Carolina and South Carolina and 3 out of 5 samples from Arkansas and Georgia tested positive for R. lauricola, whereas none of the symptomatic samples collected in Louisiana or Texas tested positive for the pathogen (Table 1). None of the samples from non-symptomatic trees gave a positive result with the LAMP assay, regardless of whether or not laurel wilt was present at the location (Tables 1 and 2).
Fungal Isolation
Results of the fungal isolations were all consistent with the results of the LAMP assays (Tables 1 and 2). Growth of R. lauricola was only observed for those samples that tested positive with the LAMP assay, while no growth was observed when the results of the LAMP assay were negative.
Objective II: In-Field Testing of the LAMP Assay
LAMP Assay on a Portable Device
The results of the in-field testing of symptomatic samples are reported in Table 3, and an example of individual runs results from each state is shown in Figure 1. The LAMP assay was capable of successfully detecting R. lauricola directly at the sites with infected trees using a field-appropriate crude extraction protocol and a Genie® III LAMP device. No amplification in the negative wells was observed, and all positive controls amplified successfully. In Georgia, 3 out of 4 redbay and 3 out of 4 symptomatic sassafras tested positive for laurel wilt disease, and in South Carolina, 3 out of 5 redbay and 3 out of 3 sassafras were positive. In North Carolina, 4 out of 6 redbay tested positive, but in both Tennessee and Kentucky, 6 out of 6 sassafras tested positive (Table 3).
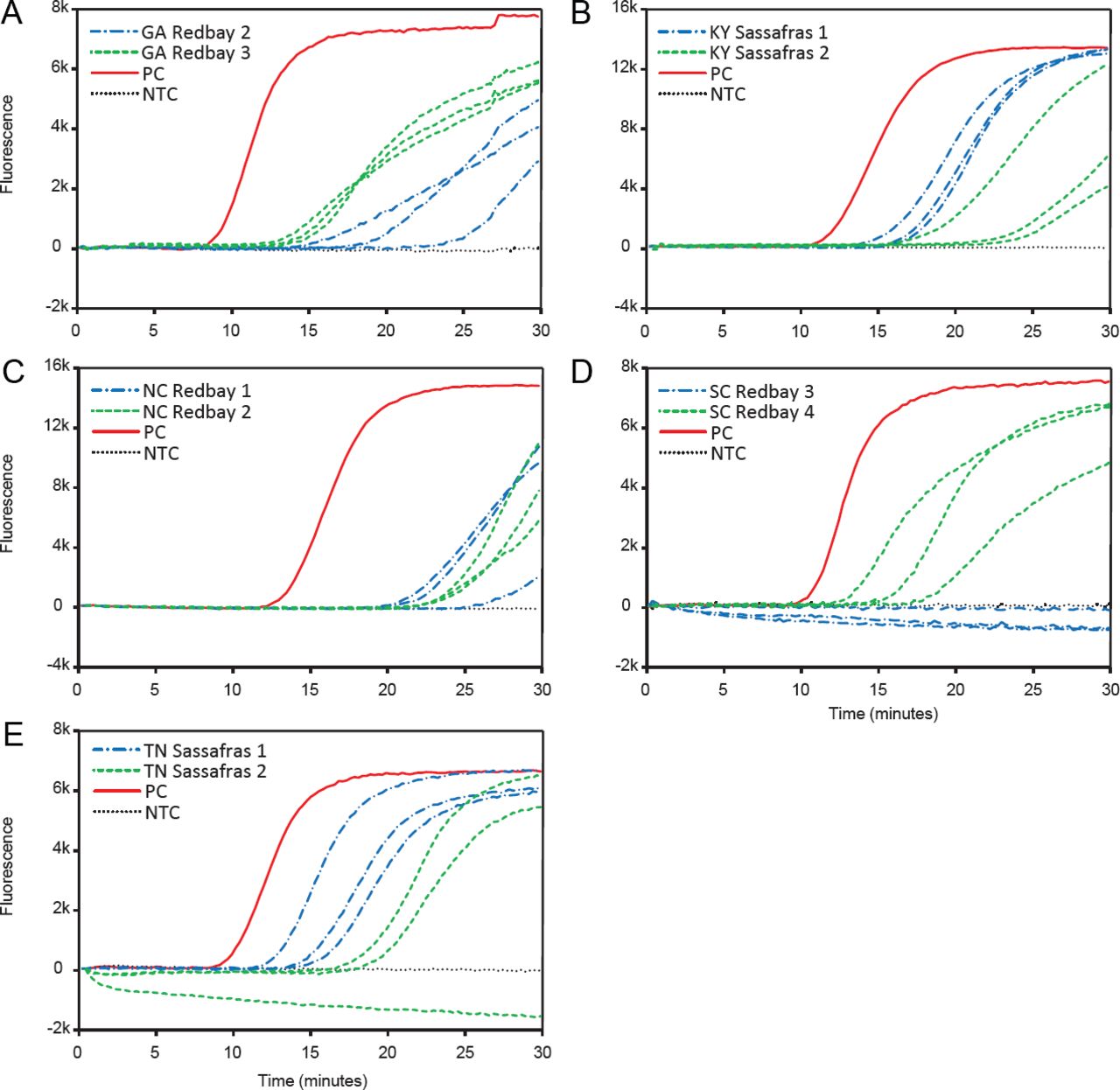
Examples of results of the in-field tests performed on a Genie® III portable LAMP device to detect Raffaelea lauricola using symptomatic samples from (A) Georgia (GA), (B) Kentucky (KY), (C) North Carolina (NC), (D) South Carolina (SC), and (E) Tennessee (TN). Positive results are identifiable by a rapidly increasing rate of fluorescence over time, indicating the successful amplification of target DNA. Template of each reaction was 1 μL of the 5% Chelex 100 crude extracted DNA. Each reaction was run in 3 replicates. NTC, no template control; PC, positive control.
Verification of Field Results
Results of the fungal isolations were mostly consistent with the results of the LAMP assay (Table 3), but discrepancies in some of the results were observed. Specifically, 2 of the sassafras samples that tested positive at the Georgia site and 1 of the sassafras samples that tested positive in the South Carolina site did not result in growth of R. lauricola once plated. High quality DNA of these wood samples was extracted in the laboratory and tested again with a LAMP assay on benchtop equipment, and a positive result was observed in all cases (Figure 2), confirming that the results previously observed in the field were a true positive. On the other hand, the 2 redbay samples that tested negative in the North Carolina site and 1 of the redbay samples that tested negative in the South Carolina site resulted in growth of R. lauricola once plated, indicating that the results previously observed in the field were a false negative. To further assess if the false negative was potentially due to inhibition of the reaction, a new crude extraction with less starting plant material was performed in the laboratory, followed by a LAMP reaction on the portable device. Results of this second reaction were all positives for the presence of R. lauricola (Table 3), confirming that the false negative results observed in the field were probably due to inhibition of the reaction. Overall true positive rate of the LAMP assay performed in field conditions was 90%.
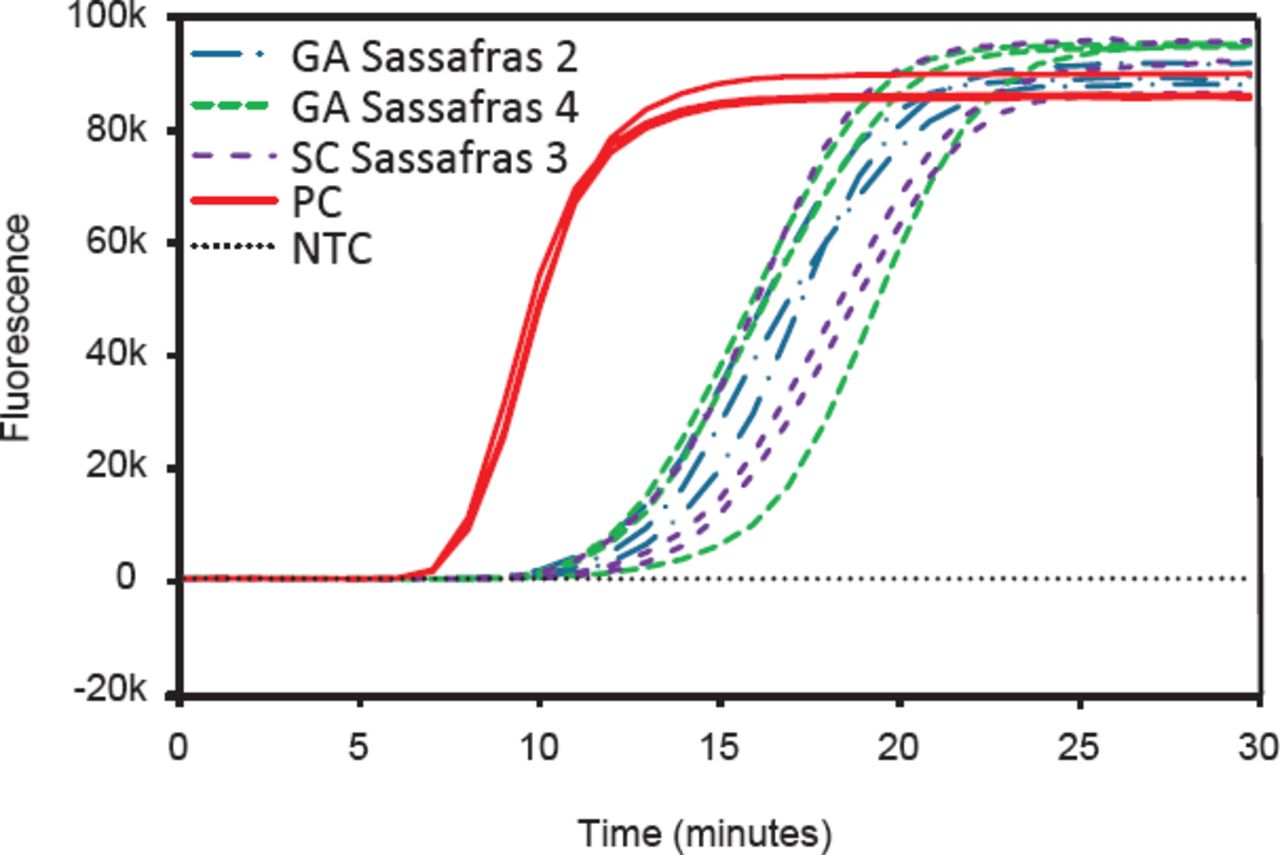
Example of a laboratory assessment of discording results between in-field LAMP assay using a portable device and fungal plating. Positive results are identifiable by a rapidly increasing rate of fluorescence over time, indicating the successful amplification of target DNA. Samples that tested positive in the field, but that did not result in growth of Raffaelea lauricola when plated in growing media, were further assessed by high quality DNA extraction followed by a LAMP assay on benchtop equipment. Each reaction was run in 3 technical replicates. GA, Georgia; SC, South Carolina; NTC, no template control; PC, positive control.
DISCUSSION
This study demonstrates that a LAMP-based molecular assay can rapidly and accurately detect the laurel wilt disease pathogen R. lauricola directly in-field without the reliance on an external laboratory. Building upon previous work that developed the species-specific LAMP primers (Hamilton et al. 2020), we were able to show that the assay is capable of correctly detecting the pathogen directly in naturally infected samples, even when multiple organisms are likely to be colonizing the tissues. Moreover, we were able to determine the reliability of the LAMP assay when conducted directly in the field on portable equipment. The actual positive rate of the in-field testing was 90%, and the test provides great advantages when one considers that the diagnostic response (which included sampling of identified trees, crude DNA extraction, and LAMP reaction) was completed within an hour of arriving on site. This is an improvement when compared to previous confirmation strategies that could take a week or more (Dreaden et al. 2014; Hughes et al. 2015) and exemplifies how the implementation of LAMP could provide a robust detection tool that could improve integrated pest management of laurel wilt disease. By providing immediate and accurate diagnoses at infection locations, management practices such as the sanitation removals of infected trees could be made without delay.
Due to the generic features of laurel wilt symptoms (Fraedrich et al. 2008), the disease can be misdiagnosed. We received several putatively infected redbay and sassafras samples collected by different personnel involved in the disease surveillance operations. All of these putatively infected samples displayed some degree of wilting and sapwood discoloration, which resembled symptoms of laurel wilt, yet many tested negative, and R. lauricola could not be isolated, illustrating how diagnosis based on visual symptoms still requires pathogen detection for disease confirmation, which to date relies on external laboratories. This is particularly important when the symptoms observed in lauraceous species are associated with colonization by different Asian ambrosia beetles, such as Xylosandrus compactus and Xylosandrus crassiusculus, which do not carry R. lauricola or similar pathogens, but can also result in sapwood discoloration and dieback of small branches (Chong et al. 2009; Ranger et al. 2016).
One of the challenges of performing point-of-care diagnostic testing is the limited control over sample quality. In our experiment, for instance, we had 3 false negatives. When samples were retested in the laboratory, however, samples tested positive, which was the correct diagnosis as confirmed by isolation of the fungus in culture. The only difference between the protocol in the field and the protocol in the lab was that in the lab we used less starting material for the extraction. This suggests that the reactions in the field failed due to the presence of inhibitors in the template. The possibility that the false negatives observed in-field were due to degradation of the reagents, on the other hand, was excluded because the positive controls amplified as expected both in terms of time and intensity. The presence of inhibitors in naturally infected, darkly pigmented samples was a common issue we encountered as we were troubleshooting the crude DNA extraction protocol. Ultimately, this is the reason we decreased the amount of wood used during extractions and reduced the volume of template per reaction from 5 μL to 1 μL for in-field LAMP tests, compared to the protocol described in Hamilton et al. (2020). Despite the presence of inhibitors, however, extracting from discolored wood tissues is still preferred, as it maximizes the probability of pathogen detection when the fungus is present.
Three of the sassafras samples that tested positive for the presence of R. lauricola, both in the field and with further assessment in the laboratory, failed to exhibit growth when plated on a media. This might be because the fungus was no longer viable, even while its DNA was still present. The in-field testing phase of this study was conducted in the autumn and winter seasons, when sassafras has already shed its leaves, which made it difficult to determine how long sampled trees had been dead. Over time, the sapwood of trees with laurel wilt dries out, and it becomes increasingly difficult to isolate R. lauricola from samples (Fraedrich 2020, unpublished data). The 3 samples in question may have come from trees that had been dead for many months, and sample moisture content and colonization by secondary organisms may have affected the ability to isolate R. lauricola. Yet, the LAMP assay was still able to detect the presence of R. lauricola DNA in samples.
Future work should focus on further optimization of in-field LAMP-based diagnostic procedures. This includes the development of commercially available lyophilized, ready-to-use reagents and a more efficient crude DNA extraction protocol, preferably without the need of a dedicated power source (for instance, as in Aglietti et al. 2019). Current LAMP technology requires minimal training for personnel performing the assay (Tomlinson et al. 2013; Thiessen et al. 2018). Additional improvements could further reduce the need for technical expertise and increase the ease and portability of the assays.
In conclusion, this study demonstrates the successful use of LAMP-based technology to rapidly diagnose laurel wilt disease and possibly other tree pathogens directly in-field. These results should foster the in-field use of this technology for other forest pathogens as well, ultimately enabling better disease management. The capability of LAMP to provide rapid and accurate in-field confirmation of forest and other tree diseases contributes to an effective early detection and rapid response system to mitigate the impact of these damaging agents (Luchi et al. 2020).
ACKNOWLEDGMENTS
We thank David Jenkins (South Carolina Forestry Commission), Chandler Barton (Arkansas Department of Agriculture, Forestry Division), Wayne Langston and Jim Slye (North Carolina Forest Service), Chip Bates and Chris Barnes (Georgia Forestry Commission), Jaesoon Hwang, Stephen Clarke, and Susan Best (USDA Forest Service), John Henderson and Rick Martin (Tennessee Forestry), Alexandra Blevins (Kentucky Division of Forestry), and Megan Buland (University of Kentucky) for their assistance in field collection and in-field testing. Funding for this project was provided by The USDA Forest Service, Forest Health Protection Special Technology Development Program (Grant #: 17DG11083150006) and the D.B. Warnell School of Forestry and Natural Resources, University of Georgia.
Footnotes
Conflicts of Interest:
The authors reported no conflicts of interest.
- © 2021, International Society of Arboriculture. All rights reserved.
LITERATURE CITED









{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.