
Abstract
The detection and identification of wood-rotting fungi in standing trees is crucial for the prediction of the severity and evolution of decay. In the case of very active root and butt rot fungi, an early identification method is important to establish the more appropriate failure risk classification. This work is aimed at reviewing the biomolecular methods recently developed to identify, directly from wood, some of the most important and widespread decay fungi. The whole method is based on the use of taxon-specific primers combined in five multiplex polymerase chain reactions (PCRs). Three multiplex PCRs allow identifying Armillaria, Ganoderma, Hericium, Inonotus, Laetiporus sulphureus, Perenniporia fraxinea, Phellinus, Pleurotus, Schizophyllum, Stereum, Trametes, and Ustulina deusta. The two remaining multiplex PCRs were developed for subgeneric identification of fungi belonging to Ganoderma, Inonotus, and Phellinus. In validation assays, multiplex PCRs allowed successfully detecting fungi in 83% of wood samples collected from decay-affected trees. Thus, the methods proved to be efficient and specific for the diagnosis and the early detection of decay fungi on standing trees.
Wood decay fungi are peculiar because of their ability to decompose lignified cell walls using enzymatic and nonenzymatic systems (Blanchette 1991). Although in forest ecosystems wood decay fungi play a positive role as primary biotic decomposers of wood logs and debris, in urban landscapes they pose serious risks by affecting the structural integrity of roots and trunks of standing trees. The loss of wood mechanical strength caused by these organisms is inherently linked to hazardous situations such as tree wind throws or limb failures, often resulting in significant damage of property and injuries. Furthermore, the establishment of decay in living trees is positively affected by urban environmental stresses that range from a general weak ening of the tree defense system to frequent injuries on branches and roots, allowing the wood-rotting agents to gain entry through wounds (Lonsdale 1999).
The detection of potentially hazardous trees is mainly based on visual tree assessment (VTA), an approach based on a visual inspection of signs and symptoms associated with anomalies in the structure of trees (Mattheck and Breloer 1992). However, VTA rarely allows the detection of incipient decay or the identification of the rotting fungi involved. Although modern technologies are improving our ability to detect internal wood decay (Tomikawa et al. 1990; Habermehl et al. 1999; Müller et al. 2001; Sambuelli et al. 2003), the identification of the agents responsible for such decay is not always feasible without the presence of fungal fruiting bodies, which are sporadically visible and frequently show overlapping characters that can lead to confusion.
Despite it all, because the biology and ecology of these fungi are variable, their identification is important to predict, to some extent, the typical pattern of spread within the tree, the effect on wood strength (Lonsdale 1999), and the risk of spread from one tree to the neighboring ones.
This is particularly important for rapidly progressing root and butt rot fungi that can turn a sound tree into a hazard in a short period of time.
Several techniques that have been used to detect incipient decay include isolation and characterization of fungal mycelia followed by the comparison of growth rates, enzymatic capabilities, and biochemical and immunologic assays (Nobles 1965; Stalpers 1978; Anselmi and Bragaloni 1992; Jellison and Jasalavich 2000; Clausen 2003). The main drawback of culture diagnosis is the difficult and time-consuming process required to obtain pure culture isolates from incipient decayed wood. Moreover, the identification of closely related taxa based on the examination of mycelial characters is often complicated and impractical in some instances. Biochemical and immunologic techniques were largely developed for the detection and identification of common brown rot fungi from lumber (Jellison and Jasalavich 2000; Clausen 2003). Although such methods do not always require pure culture isolation, they lack sensitivity for detection of fungi in the early stage of infection.
As already reported (Guglielmo et al. 2007), the techniques based on fungal DNA detection are a promising alternative for specific, sensitive, and rapid routine diagnoses directly from wood samples. Polymerase chain reaction (PCR) methods based on taxon-specific primers designed on nuclear or mitochondrial ribosomal DNA (rDNA) loci proved to be effective for the identification of fungi at different taxonomic levels (Moreth and Schmidt 2001; Bahnweg et al. 2002; Gonthier et al. 2003). Furthermore, multicopy arrangement and highly conserved priming sites, typical of both nuclear and mitochondrial rDNA, permit amplification from virtually all fungi, even if the starting sample is lacking in quantity or quality such as DNA extracts directly from wood (Jasalavich et al. 2000). As an added benefit, the simultaneous application of taxon-specific primers in multiplex PCR reactions has been reported in clinical and food microbiology to increase the diagnostic capacity of PCR (Elnifro et al. 2000; Corbiere Morot-Bizot et al. 2004).
The objective of this study was to develop a multiplex PCR-based method for the identification of some of the most important wood-rotting fungi in standing trees. This article also reports on the validation of such methods on wood samples.
MATERIALS AND METHODS
Target Decay Fungi, Sampling, and Culturing
We selected the target fungi based on their frequency in the northern temperate areas and their aggressiveness (Hickman and Perry 1997; Lonsdale 1999; Nicolotti et al. 2004; Bernicchia 2005). Selected taxa included Armillaria spp., Ganoderma spp., Hericium spp., Inonotus spp., Laetiporus sulphureus, Perenniporia fraxinea, Phellinus spp., Pleurotus spp., Schizophyllum spp., Stereum spp., Trametes spp., and Ustulina deusta. The design and the initial validation of taxon-specific diagnostic PCR assays were based on 80 fungal collections, either pure cultures or fruiting bodies specimens, encompassing the most representative species per each taxon (Table 1).
Multiplex polymerase chain reaction (PCR) primer combinations, diagnostic purposes, and positively tested species.
Fungal collections were either provided by CABI Bioscience National Center of Wood Rotting Fungi–Garston (U.K.), CAS Institute of Microbiology–Videnska (CR), METLA Finnish Forest Research Institute-Vantaa (FIN), MUAF Faculty of Forestry and Wood Technology–Brn o (CR), USDA Forest Products Laboratory–Madison (WI, U.S.), USDA Agricultural Service–Davis (CA, U.S.) and WSL Swiss Federal Research Institute–Birmensdorf (CH) or obtained from fruiting bodies collected in California and Italy and identified through analytical keys (Hickman and Perry 1997; Bernicchia 2005). Pure fungal cultures were isolated from the context of the fruiting bodies by using potato dextrose agar (Merck KGaA, Darmstadt, Germany) medium with 0.2 g/L (1.8 10−3 oz/gal) of streptomycin sulphate. Before DNA extraction, isolates were subcultured in a 2% (w/v) liquid malt extract (AppliChem GmbH, Darmstadt, Germany) medium for approximately 2 weeks at room temperature. Cultures were harvested by filtration and lyophilized.
Taxon-Specific Primers Design and Testing
DNA was extracted from lyophilized mycelia or dried fruiting bodies specimens using a modified cetyltrimethylammonium bromide extraction method (Hayden et al. 2004).
Ribosomal DNA amplifications of the 5′ end portion of the nuclear large subunit and of the portion including the internal transcribed spacers (ITSI and ITSII) were performed using the fungal-specific primers pairs ctb6 and tw13 (White et al. 1990; O’Donnell 1993) and ITS1-F and ITS4 (White et al. 1990; Gardes and Bruns 1993), respectively. For taxa lacking suitable portions for taxon - specific primer design in the previously mentioned regions, a portion of mitochondrial small subunit was amplified using primers MS1 and MS2 (White et al. 1990).
PCR products were cleaned by using Qia-quick purification kit (Qiagen, Valencia, CA) and cycle-sequenced with a BigDye Terminator v. 3.1 cycle-sequencing kit (Applied Biosystems, Foster City, CA). Sequencing reactions were loaded on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA).
Additional sequences available in the European Bioinformatics Institute nucleotide sequences database (EMBL-EBI; www.ebi.ac.uk) were used to increase the sample size of target taxa and ensure specificity of primers by including as many representatives as possible of the target taxon and closely related nontarget taxa. Domains conserved within a taxon, but variable among taxa, were chosen as target regions for taxon-specific primer design. To identify these regions, all sequences from a given taxon were aligned using CLUSTALW (Thompson et al. 1994) with one sequence of a closely related species used for outgroup comparison.
Taxon-specific primers to be used in multiplex PCR were designed on the selected regions using the software PRIMER3 (www.genome.wi.mit.edu/cgi-bin/primer/primer3) to be highly specific for the target fungal taxa. The annealing temperature of each primer pair was optimized using a thermocycling gradient to improve PCR efficiency with the highest stringency.
Multiplex Polymerase Chain Reaction Development
Multiplex PCR was performed by combining taxon-specific reverse primers with similar annealing temperatures and variable amplicon sizes (Guglielmo et al. 2007). Amplified DNA fragments were visualized on agarose gel.
Validation of the Polymerase Chain Reaction-Based Method on Field Samples
Wood samples from trees with evident decay symptoms were analyzed to test the sensitivity and reliability of the method. Sampled trees belonged to 19 species and 15 genera, including Acer, Aesculus, Cedrus, Celtis, Fagus, Juglans, Malus, Platanus, Populus, Prunus, Quercus, Robinia, Sophora, Tilia, and Ulmus. Wood samples were obtained either through a Swedish increment borer near the point of fruiting body emergence or through a scalpel where incipient decay was visible.
Wood samples, approximately 150 mg (5.25 10−3 oz fresh weight) each, were lyophilized, homogenized through a FastPrep FP120 Cell Disrupter (Qbiogene, Irvine, CA) and extracted with the QIAamp DNA Stool Mini Kit (Qiagen). Each DNA extract was diluted 100-fold and amplified through the multiplex PCR-based method.
The expected fungal taxon per each sample was assessed either through the analysis of macroscopic features of the fruiting bodies or through BLAST search analysis of the sequenced ITS region amplified with the primers pair ITS1-F and ITS4. The diagnostic efficiency of the method was determined by evaluating its ability to detect and correctly identify the fungal taxon expected per each sample (Hickman and Perry 1997; Bernicchia 2005).
RESULTS
Taxon-Specific Priming Multiplex Polymerase Chain Reactions
Taxon-specific primers (Guglielmo et al. 2007, 2008) were combined in five multiplex PCR reactions (Table 1). Taxon-specific primer for Ustulina deusta was designed for this study (5′-GCTCATCTCTACAGGCGAGAA-3′). The universal fungal primers ITS1-F and ITS4 were used in one multiplex (M1) to evaluate the efficiency of fungal DNA extraction, avoiding, thus, possible false-negatives resulting from either undetectable DNA quantities or PCR inhibitory compounds. M1 also allows identification of fungi belonging to the genera Ganoderma and Inonotus or Phellinus. M2 allows identifying Armillaria, Hericium, Laetiporus sulphureus, and Pleurotus, M3 was suitable for the detection of Perenniporia fraxinea, Schizophyllum, Stereum, Trametes, and Ustulina deusta. Mhyme and Mgano have been developed to identify at a subgeneric rank taxa belonging to the Inonotus – Phellinus group and Ganoderma, respectively.
Using the optimized reaction parameters, each multiplex PCR allowed to efficiently amplify the corresponding target taxon DNA fragment without crossreacting with nontarget DNA or producing any ambiguous or extra amplicons (data not shown).
The amplified fragments in multiplex reactions were easily differentiated and scored according to the size using gel containing 1% (w/v) of high-resolution MetaPhor and 1% (w/v) of standard agarose after a 2 hr or 2 hr 30 min electrophoretic migration at 4 V/cm (1.6 V/in) depending on the multiplex PCR (Figures 1 and 2).
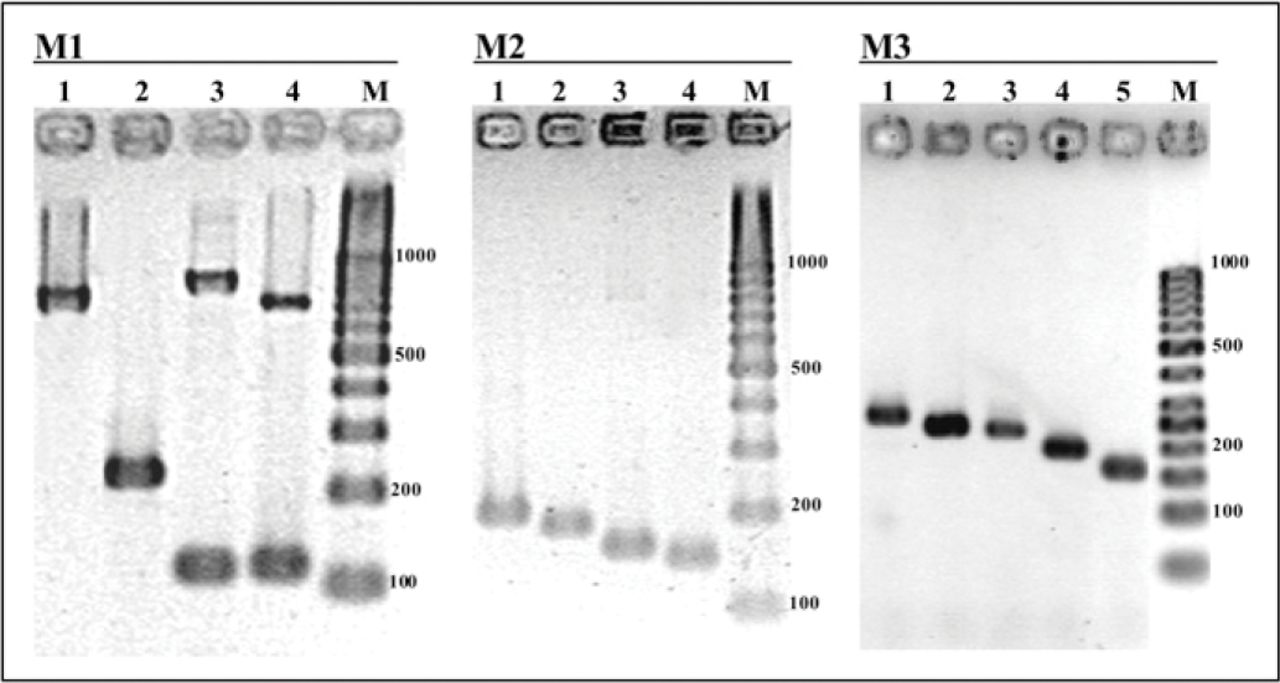
The results of M1, M2, and M3 visualized on an ultraviolet gel documentation system after 2 hr electrophoresis at 4 V/cm on a 1% Metaphor 1% standard agarose gel. M1 = Polymerase chain reaction (PCR) products from DNA extracts of Trametes versicolor (lane 1), Ganoderma adspersum (lane 2), Inonotus hispidus (lane 3), and Phellinus torulosus (lane 4), M2: PCR products from DNA extracts of Hericium erinaceum (lane 1), Armillaria mellea (lane 2), Pleurotus ostreatus (lane 3), and Laetiporus sulphureus (lane 4). M3: PCR products from DNA extracts of Ustulina deusta (lane 1), Stereum spp. (lane 2), Trametes spp. (lane 3), Schizophyllum spp. (lane 4), and Perenniporia fraxinea (lane 5). M = molecular weight marker 100 bp DNA ladder. Molecular weights are indicated in base pairs (bp).
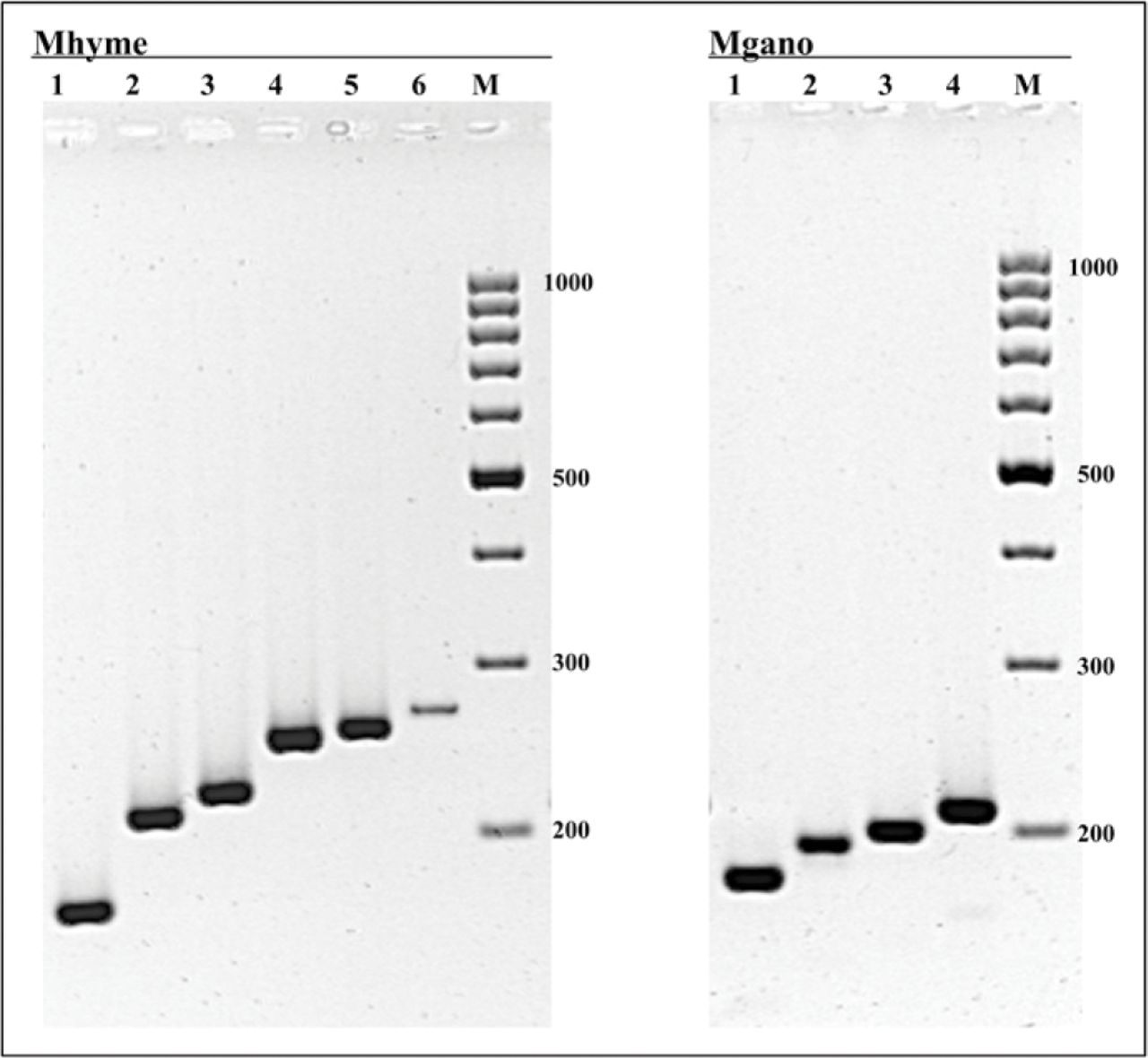
The results of Mhyme and Mgano visualized on an ultraviolet gel documentation system after 2 hr 30 min electrophoresis at 4 V/cm on a 1% Metaphor 1% standard agarose gel. Mhyme: Polymerase chain reaction (PCR) products from DNA extracts of Phellinus tuberculosus (lane 1), Inonotus hispidus (lane 2), Phellinus gilvus (lane 3), Inonotus dryadeus (lane 4), Phellinus punctatus (lane 5), and Inonotus dryophilus (lane 6). Mgano: PCR products from DNA extracts of Ganoderma resinaceum (lane 1), Ganoderma lucidum (lane 2), Ganoderma applanatum (lane 3), and Ganoderma adspersum (lane 4). M = molecular weight marker 100 bp DNA ladder. Molecular weights are indicated in base pairs (bp).
Validation of the Polymerase Chain Reaction-Based Method on Field Samples
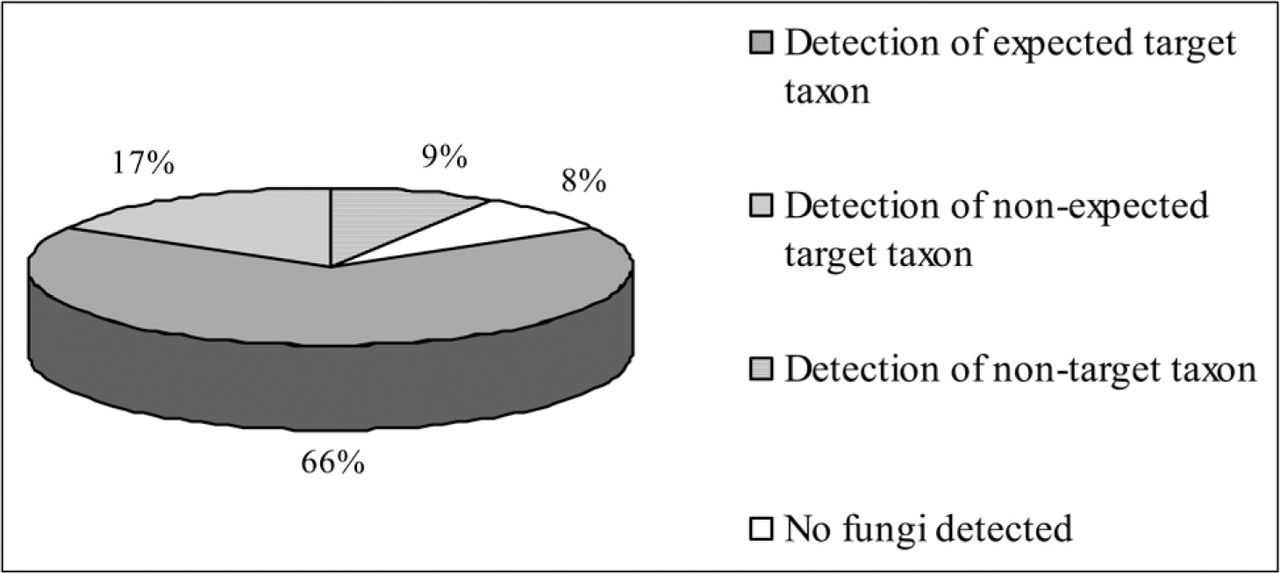
By using the PCR-based method developed in this study (Figure 3), in 83% of the samples, at least one of the target decay fungi was detected (Figure 4). In 66% of the samples, we found consistent matches between multiplex PCR-based results and the expected data. In 17% of the samples, an unexpected target decay fungi was found. Among the unexpected target taxa detected through the PCR-based method, the most common were P. fraxinea instead of Ganoderma spp. and P. torulosus and Stereum spp. instead of the Inonotus–Phellinus group. In 9% of the samples, other nontarget taxa corresponding, after BLAST search analysis, to anamorphic ascomycetes were detected.

Summary of the steps included in the method. Groups detectable by Mhyme refer to taxonomic groups described by Wagner and Fischer (2001, 2002).

Diagnostic efficiency of the method on wood samples taken from decay-affected trees. For details, see the text.
DISCUSSION
The PCR-based method developed in the current study allowed detecting and identifying decay fungi deemed to be responsible for the majority of urban tree failures in the north temperate areas of the world. Moreover, this molecular method can be applied on fungal DNA extracted directly from wood, allowing, thus, to bypass the time-consuming and not always feasible fungal isolation step.
Starting from a 1/100 dilution of wood DNA extract solution obtained by using the QIAamp DNA Stool Mini Kit™ on approximately 150 mg (5.25 10−3 oz) of fresh wood, the M1 is performed to detect whether any fungal DNA is present in the sample as well as to identify species belonging to Ganoderma and/or to Inonotus – Phellinus group (Figure 3). No amplification of any DNA fragments after this reaction may indicate either no successful purification of fungal DNA from PCR inhibitory compounds present in the wood such as polyphenols, tannins, and resin acids or the absence of fungi at a detectable concentration in the sample. In this case, it is advisable to repeat the multiplex PCR using a different DNA dilution. If the negative result is still confirmed, a further DNA extraction from wood may be required. Conversely, in the case of positive outcomes after M1, the M2 and M3 assays should be performed to detect DNA of one or more of the remaining target fungi. Furthermore, Mgano and Mhyme may be performed in the case of occurrence, after M1, of Ganoderma spp. and of Inonotus spp.– Phellinus spp.-specific amplicons, respectively. The absence of taxon-specific PCR products in any multiplex PCR, and the simultaneous presence of the amplified DNA fragment corresponding to the ITS region in M1, may indicate the detection of a nontarget fungal taxon. In this case, it could be advisable to sequence the PCR product to ascertain, after BLAST search analysis, what fungus is involved.
Species-level diagnostic primers were developed for three fungal taxa frequently reported for their important role in wood decay processes: L. sulphureus, P. fraxinea, and U. deusta (Lonsdale 1999; Nicolotti et al. 2004). Likewise, Mhyme and the Mgano were developed to identify at a subgeneric rank the most hazardous taxa within these genera and to differentiate them from fungal species known to be less hazardous. Mhyme-specific primers allowed to identify, at the level of the groups described in the classification of Hymenochaetales re-examined by Wagner and Fischer (2001, 2002), very active decay species that can seriously affect the stability of trees in urban landscape such as I. dryadeus, I. hispidus, P. gilvus, and P. robustus. Through Mgano, European G. lucidum, reported as a slow white rot fungus of many broadleaved trees rarely associated with their failures (Bernicchia 2005), is well distinguishable from other Ganoderma species such as G. adspersum, G. applanatum, and G. resinaceum known to significantly affect tree stability (Lonsdale 1999; Bernicchia 2005). Moreover, G. lucidum from North America, reported to cause rapid and extensive rot in Quercus spp. and Acer spp. (Hickman and Perry 1997; Dreistadt et al. 2004), is identified, through Mgano, as a G. resinaceum group, as already inferred by a phylogenetic study based on small-subunit ribosomal DNA sequences (Hong and Jung 2004).
Validation assay on field samples highlights the specificity and efficiency of the PCR-based method, as suggested by the consistent matches between PCR-based results and the expected data. The occurrence of unexpected fungi were attributable either to visual misidentification of the emerging fruiting bodies such as in the case of P. fraxinea incorrectly identified as P. torulosus or the association between different fungal taxa in a same tree as reported in this study with Ganoderma spp. and P. fraxinea. In this case, patchy patterns of wood localization of decay fungi may allow the detection of only one of the different fungi involved depending on the wood sample.
PCR-based identification proved to be efficient starting from decayed wood samples taken at the point of fruiting body emergence as well as from wood core samples extracted with a Swedish increment borer. Such results suggest that this PCR-based method may be used by arborists as a complement to VTA analysis. The method described here can be easily performed in disease diagnostic clinics equipped with basic molecular biology instruments. Further studies, aimed at setting up and standardizing field samples, are in progress.
- © 2009, International Society of Arboriculture. All rights reserved.
LITERATURE CITED









{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.